Anthropic explora tornar o Claude um químico
Anthropic detalha como o Claude começa a ler estruturas, espectros e literatura química, já rivalizando com softwares de NMR em tarefas práticas e apontando rotas para P&D.
Danilo Gato
Autor
Introdução
Em 5 de junho de 2026, a Anthropic publicou um estudo mostrando que o Claude, em sua versão Opus 4.7, começa a atuar como um verdadeiro assistente químico. A proposta é clara, transformar o modelo em suporte prático para análises como previsão de NMR e elucidação estrutural, tarefas que consomem tempo em laboratórios e equipes de P&D. A palavra chave aqui é Claude químico, não um hype, mas um conjunto de resultados testados contra ferramentas tradicionais.
A importância é direta para quem desenvolve fármacos, agrotinsumos, polímeros ou materiais funcionais. Ler espectros, ligar picos a átomos, reconciliar dados de massa com fórmulas, tudo isso é rotina. A Anthropic afirma que o Claude já ajuda a traduzir representações, do SMILES a figuras de artigos, e a integrar leituras de espectros e métodos experimentais, algo que até pouco tempo exigia softwares especializados e forte supervisão humana.
O artigo cobre três dimensões práticas, o que o Opus 4.7 trouxe de novo para química, o desempenho em NMR frente a ChemDraw e MestReNova e como aplicar com responsabilidade em fluxos de P&D, com ganhos reais sem ignorar limitações.
Por que transformar o Claude em um químico importa
Química é linguagem múltipla. Um mesmo composto aparece como desenho em figuras de artigos, como SMILES em bancos de dados, como nome sistemático em patentes e como listas de picos em espectros. Traduzir entre essas camadas é trabalho de formiguinha, especialmente quando o volume de literatura cresce rápido. A CAS indica mais de 290 milhões de substâncias no CAS REGISTRY, com novas entradas adicionadas continuamente a partir de literatura e solicitações, o que pressiona fluxos de busca, triagem e verificação.
Modelos multimodais com capacidade de raciocínio explícito começam a mudar o quadro. A Anthropic argumenta que o Claude já lê estruturas em figuras e métodos experimentais como eles aparecem no paper, não só em bases pré processadas, além de expor o raciocínio passo a passo, o que permite auditoria por parte do químico. Essa combinação mantém o especialista no controle, mas transfere parte do fardo operacional para a IA.
Na prática, isso significa acelerar tarefas de confirmação estrutural, triagem de candidatos e preparação de relatórios. Em ambientes regulados, manter rastreabilidade e revisão crítica continua obrigatório, mas contar com um copiloto que acerta a maior parte do trabalho braçal libera tempo para decisões de rota sintética, seleção de condições e desenho de estudos complementares.
O que há de novo no Claude Opus 4.7 para química
O Opus 4.7 é a geração do Claude com melhorias de entendimento multimodal e raciocínio em tarefas longas, com documentação oficial citando ganhos na leitura de diagramas técnicos e estruturas químicas. Em paralelo, a Anthropic e parceiros distribuem o modelo via plataformas como Amazon Bedrock, o que facilita integrar o Claude em pipelines que já rodam em nuvem corporativa. Para uso prático, disponibilidade e latência importam tanto quanto métricas.
Embora a página de produto e os posts de anúncio foquem em benchmarks gerais, o estudo de 5 de junho detalha o foco em química. A pesquisa avalia o Opus 4.7 contra ChemDraw 25.0.2 e MestReNova 17.0.0 em duas direções, previsão de deslocamentos e padrões a partir de uma estrutura, e elucidação estrutural a partir de espectros 1D de ¹H e ¹³C mais HRMS. Esse desenho aproxima a avaliação do que acontece em bancada, onde o espectro chega primeiro e a dúvida estrutural vem logo depois.
Do ponto de vista de adoção, o fato de o Claude operar a partir de insumos que um químico literalmente cola em um chat, fórmulas, listas de picos e solventes, reduz a fricção. Não requer instalação, arquivos proprietários ou treinamento longo de software, e ainda expõe raciocínio para conferência.
Resultados em NMR, comparação com ChemDraw e MestReNova
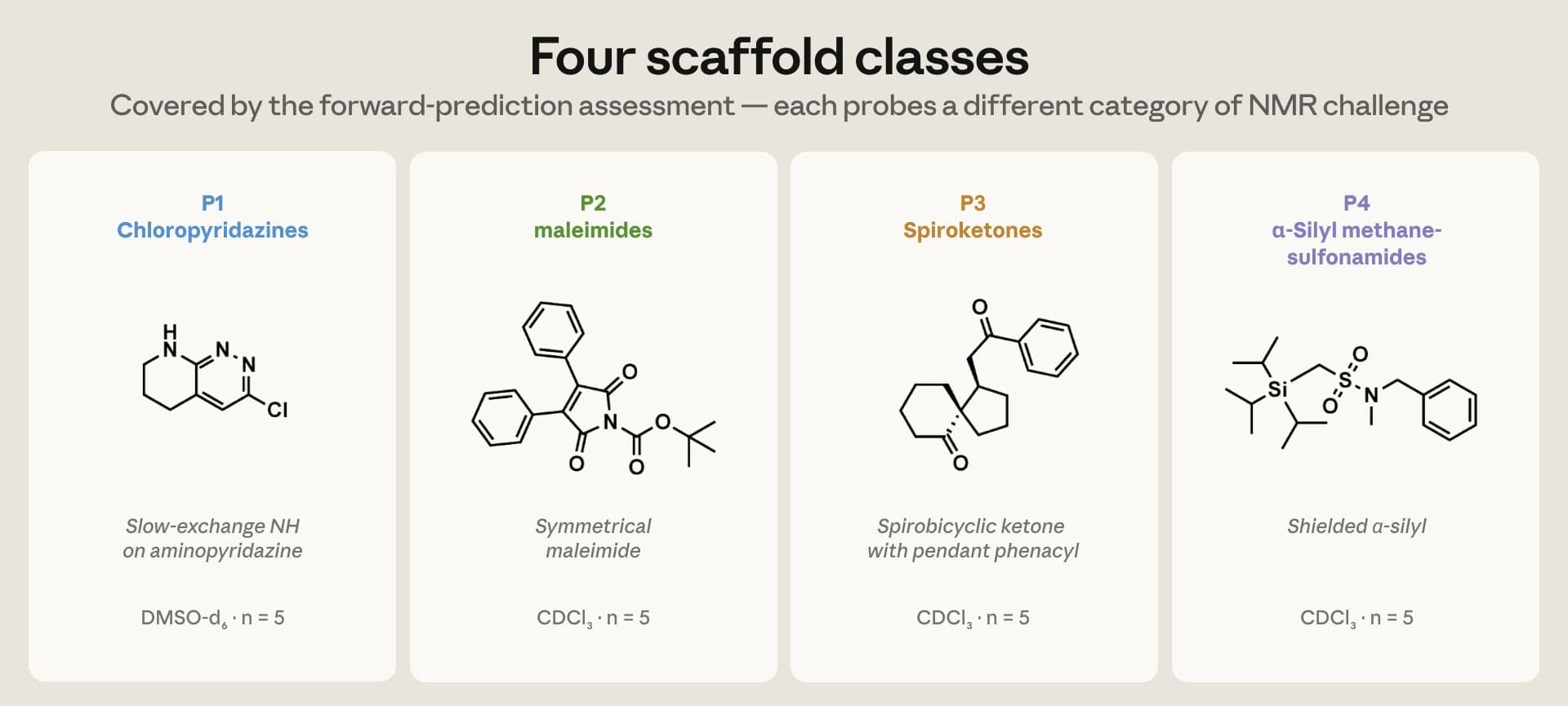
O conjunto de teste inclui 20 compostos de preprints do ChemRxiv para a tarefa direta, estrutura para NMR, cobrindo quatro famílias com desafios diferentes, e 15 alvos para a tarefa inversa, NMR e HRMS para estrutura. A seleção foi feita antes de rodar previsões, um ponto importante contra viés de escolha.
Nos testes de previsão de deslocamento, ¹H e ¹³C, o Opus 4.7 teve o menor erro médio em ¹H, MAE de 0,079 ppm, e resultado comparável ao MestReNova em ¹³C, 1,37 ppm contra 1,48 ppm, mantendo também boa consistência entre réplicas. No que diz respeito a multiplicidade e J acoplamentos, os modelos do Claude superaram ChemDraw e MestReNova, alcançando cerca de 80 a 84 por cento dos pares dentro de 0,5 Hz, contra 26 a 35 por cento nos softwares clássicos.
![Resumo de erro e cobertura em NMR, Claude vs. softwares clássicos]
Na elucidação estrutural a partir de espectros 1D e HRMS, a tarefa realmente crítica quando se precisa confirmar produto de reação, o Opus 4.7 recuperou todos os oito alvos mais simples em todas as tentativas, e nos sete mais densos, ao receber somente o SMILES do material de partida como dica extra, retornou o acerto em todas as rodadas para parte deles e em duas de três para os restantes. Esses resultados estendem a automação para uma zona onde, no cotidiano, recorre-se a 2D NMR ou interpretação manual.
![Painel de acertos na tarefa de elucidação estrutural]
Por que isso importa no chão de fábrica ou no laboratório de descoberta, porque encurta o ciclo confirmar produto, ajustar rota, avançar lote. Quando o modelo acerta multiplicidade e J com precisão, fica mais rápido concluir se uma impureza é relevante, se há sinal de subproduto esperado ou se uma etapa precisa de reprojeto. Ainda assim, manter a dupla conferência com um software determinístico ajuda a flagrar vieses comuns, como o deslocamento sistemático de carbonilas um pouco abaixo do experimental, observado tanto nos modelos quanto em ferramentas clássicas.
Limitações, riscos e onde o Claude ainda erra
A amostra do estudo é pequena, 20 compostos para previsão direta e 15 para elucidação, e foca em 1D NMR com solventes específicos, DMSO d6, CDCl3 e D2O. Não cobre 2D, estereoquímica ou solventes como metanol d4, benzeno d6 e acetona d6. Em alguns alvos mais densos, sem a informação do material de partida, o modelo pode circular no raciocínio sem cravar uma estrutura final. Esses recortes importam para evitar extrapolações indevidas.

Outra limitação prática, ferramentas tradicionais como MestReNova e ChemDraw são determinísticas e amplamente conhecidas, com manuais, cursos e validações internas. Quando se integra um LLM, entram fatores como variabilidade entre rodadas e sensibilidade a prompt. O estudo mitigou isso rodando réplicas e reportando faixas, mas equipes devem refletir essas boas práticas, rodar mais de uma semente, comparar contra curvas mestre e manter protocolos de aceitação de resultado.
Por fim, apesar do entusiasmo, a recepção a modelos de linguagem em produção varia. Em comunidades técnicas surgem relatos positivos e negativos, e a mensagem que fica é calibrar o uso ao contexto, medir com dados da sua aplicação e manter plano de fallback. A própria disponibilidade via Bedrock ajuda a operacionalizar testes A B e governança.
Aplicações práticas em P D farmacêutico e materiais
Três trilhas de uso ganham tração imediata. Primeiro, confirmação estrutural rápida em química medicinal. Cole listas de picos ¹H e ¹³C, fórmula exata de HRMS e solvente, peça ao Claude para propor correspondências pico átomo e sugerir estruturas candidatas. Valide contra uma previsão determinística no ChemDraw ou no MestReNova e aceite apenas quando houver concordância dentro de janelas definidas, por exemplo, 0,20 ppm para ¹H e 1,0 ppm para ¹³C, replicando o critério do estudo.
Segundo, triagem de bibliografia e patentes. Use o Claude para extrair SMILES de figuras, normalizar nomes e conectar com buscas em bases internas. O ganho vem de reunir o que estaria espalhado em PDFs e anexos. Como o modelo lê diagramas e métodos como aparecem no paper, reduz o retrabalho de transcrição. Mantendo logs e revisão por pares, a equipe de P D consegue aumentar a cobertura sem inflar o tempo de ciclo.
Terceiro, planejamento sintético e revisão de mecanismos. Ainda que o estudo não tenha benchmarkado retrosíntese de forma extensiva, a Anthropic lista mecanismos, setas de elétrons e discussão de estados de transição como áreas de foco. Ao aplicar, trate o Claude como interlocutor crítico, peça hipóteses, peça contraexemplos, e valide com heurísticas do time. O objetivo não é terceirizar julgamento, e sim ampliar o leque de alternativas consideradas mais cedo.
Como começar, fluxos de trabalho e boas práticas
- Dados padronizados. Alimente o modelo com a fórmula exata, listas de picos tabuladas por núcleo e solvente, e detalhe condições quando disponíveis. Replicar o formato do estudo ajuda a estabilizar respostas e facilita comparação com previsões determinísticas.
- Réplicas e aceitação. Rode o mesmo prompt três vezes e use métricas de aceitação por janela de erro, além de uma regra de empate com ferramentas clássicas. Reserve a elucidação automática para triagem e leve ao químico a versão final.
- Auditoria do raciocínio. Exija que o Claude exponha o caminho, mapeando cada pico a um átomo proposto e justificando deslocamentos e multiplicidades com ambiente químico, por exemplo, desshielding por carbonila vizinha, acoplamentos vicinais aromáticos em torno de 7 Hz. Essa transparência é o filtro que separa acerto robusto de chute convincente.
- Integração operacional. Se a organização usa AWS, experimente o Opus 4.7 via Bedrock, o que facilita versionamento, limites, logs e isolamento de dados. Monte playbooks com prompts padrão por classe de tarefa para reduzir variabilidade entre usuários.
- Treinamento interno. Capacite o time em princípios de prompting e em leitura crítica de saídas, reforçando que o modelo é assistente. Fixe uma política onde toda decisão química relevante requer validação cruzada antes de seguir para scale up ou reporte externo.
Casos ilustrativos a partir do estudo
- Deslocamentos em ¹H. Nas séries de cloropiridazinas, um próton NH de troca lenta aparece experimentalmente entre 6,8 e 7,9 ppm. O Opus 4.7 tendeu a prever um valor um pouco abaixo, mas de forma consistente, já o Sonnet 4.6 posicionou entre 10 e 13 ppm, bem fora da faixa. Esse padrão mostra como vieses sistemáticos podem ser reconhecidos e corrigidos no fluxo.
- Multiplicidade e J. O Claude acertou mais frequentemente padrões de splitting e aproximou J com erro médio em torno de 0,5 Hz, contra cerca de 2 Hz nos softwares clássicos nos conjuntos avaliados. Em triagens rápidas, isso ajuda a notar vizinhança aromática característica e acoplamentos geminais anômalos.
- Cobertura. O ChemDraw destacou-se pela cobertura de picos, ainda que com menor precisão em alguns cenários, o que sugere política de duas ferramentas, usar o Claude para precisão e o ChemDraw para garantir que nenhuma região relevante ficou sem previsão.
Reflexões e insights
Do ponto de vista estratégico, a janela que se abre não é só de produtividade, é de aprendizado coletivo. Um assistente que explica por que associou um pico a um carbono específico, cita efeito anisotrópico e sugere alternativas, treina a equipe no processo. Esse efeito colateral de aprendizado é valioso quando o turnover é alto e quando projetos correm em paralelo.
Outro ponto, o elo entre IA geral e ferramentas de domínio ficou mais curto. O Opus 4.7 não é um modelo treinado só para NMR. Mesmo assim, ao ler figuras, SMILES e listas de picos como estão no paper, alcança paridade média com softwares especializados em partes da tarefa e leva vantagem onde o raciocínio contextual ajuda. Em escala, isso tende a viabilizar QA inteligente, revisão de cadernos e checagens automáticas entre lotes.
Por fim, não é o fim do software clássico. Pelo contrário. A combinação de LLM mais ferramentas determinísticas parece o melhor dos dois mundos, velocidade e contexto mais repetibilidade e cobertura. Com governança e métricas claras, times podem acelerar sem abrir mão de conformidade e confiabilidade.
Conclusão
O recado da Anthropic é pragmático. O Claude químico já rende valor em tarefas de NMR e começa a encurtar etapas de confirmação e triagem, principalmente graças ao Opus 4.7. As métricas do estudo são animadoras, especialmente em multiplicidade e J, e a capacidade de operar a partir de dados que um químico já tem em mãos facilita a adoção. Integrado com boas práticas, o ganho chega sem atritos desnecessários.
Seguir adiante significa ampliar o escopo, mais famílias estruturais, solventes e 2D NMR, e estabelecer padrões de uso, réplicas, aceitação e auditoria. A combinação de IA e ferramentas clássicas cria um ciclo virtuoso, mais velocidade, mais qualidade e mais clareza de raciocínio. Esse é o tipo de avanço que não promete milagres, mas que muda a rotina de quem precisa entregar moléculas corretas, no prazo e com confiança.